El Alzheimer (EA) es una enfermedad neurodegenerativa que causa deficiencias progresivas en la memoria, el lenguaje y el pensamiento, con la eventual pérdida de la capacidad para realizar actividades sociales y funcionales en la vida diaria. En general, la supervivencia promedio es de 4 a 8 años después de su diagnóstico. Existe una necesidad médica urgente e insatisfecha de tratamientos efectivos que retrasen, detengan, reviertan, prevengan o curen la enfermedad.
Aducanumab-avwa (IND 106230) es un anticuerpo monoclonal de inmunoglobulina gamma 1 (IgG1) humana recombinante, fabricado por la compañía Biogen y dirigido a formas agregadas solubles e insolubles de péptido beta amiloide (Aβ). Los depósitos extracelulares de Aβ, denominados placas amiloides, son una de las características patológicas de la EA, junto con los agregados intracelulares de proteína tau hiperfosforilada en forma de ovillos neurofibrilares. Se ha propuesto que la acumulación de Aβ en el cerebro es el principal impulsor del proceso de la enfermedad y precede a la acumulación patológica de proteína tau hiperfosforilada y degeneración neuronal.
La biotecnológica Biogen presentó a la FDA la solicitud de evaluación acelerada de su terapia biológica (BLA, del inglés Biologics License Application) a fecha 7 de julio de 2020, y recibió la aprobación el día 6 de julio de 2021. Esta decisión ha hecho que la Agencia americana se lleve un aluvión de críticas fundamentadas en la inconsistencia de los resultados de los ensayos clínicos presentados, lo cual ha tenido como consecuencia la dimisión de varios de los consejeros externos de la FDA tras la votación.
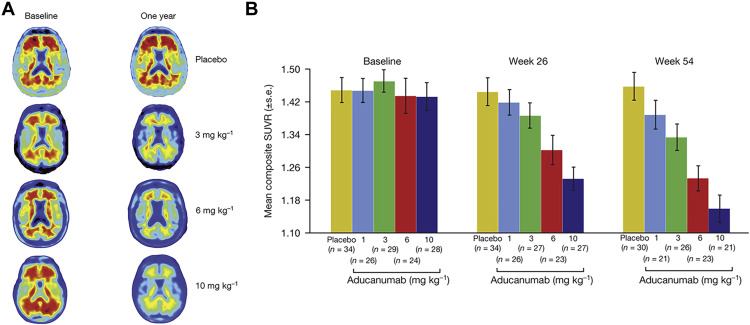
 Fuente: revisión clínica hecha por la FDA en la que se fundamentó la toma de la decisión
Fuente: revisión clínica hecha por la FDA en la que se fundamentó la toma de la decisión
Según informes de evaluadores externos de la FDA, existen discordancias en los resultados de los estudios 301 y 302 que no dejan clara la eficacia del aducanumab en el tratamiento de la EA.
Los consejeros de la FDA afirmaron que el ensayo clínico 301 es un estudio negativo que no contribuye a la evidencia de la eficacia clínica del aducanumab, sin embargo, la FDA argumenta que sus resultados pueden ser complementarios a los del estudio 302, el cual tuvo un fin prematuro.
Ante el hecho de la temprana finalización del ensayo clínico 302 la FDA consideró que los datos, aún así, eran lo suficientemente interpretables y capaces de dar evidencia de la eficacia del medicamento. La Agencia americana calificó a este estudio como “robusto y excepcionalmente persuasivo que otorga la evidencia primaria suficiente como para apoyar a la aprobación del fármaco”.
Con respecto a la concesión de la evaluación acelerada por la Agencia americana se señaló que:
«…EL EFECTO DEPENDIENTE DEL TIEMPO Y DE LA DOSIS CUMPLEN CON EL ESTÁNDAR DE EVIDENCIA SUSTANCIAL DE EFECTIVIDAD»
«…LA EVIDENCIA QUE RESPALDA EL BENEFICIO CLÍNICO ES FUERTE, PERO ESTÁ ASOCIADA A LA INCERTIDUMBRE RESIDUAL TRANSMITIDA POR LOS RESULTADOS DEL PUNTO FINAL DEL ESTUDIO 301 (Y LA CONTRIBUCIÓN ASOCIADA DE ESOS RESULTADOS A LA TERMINACIÓN PREMATURA DE AMBOS ESTUDIOS)»
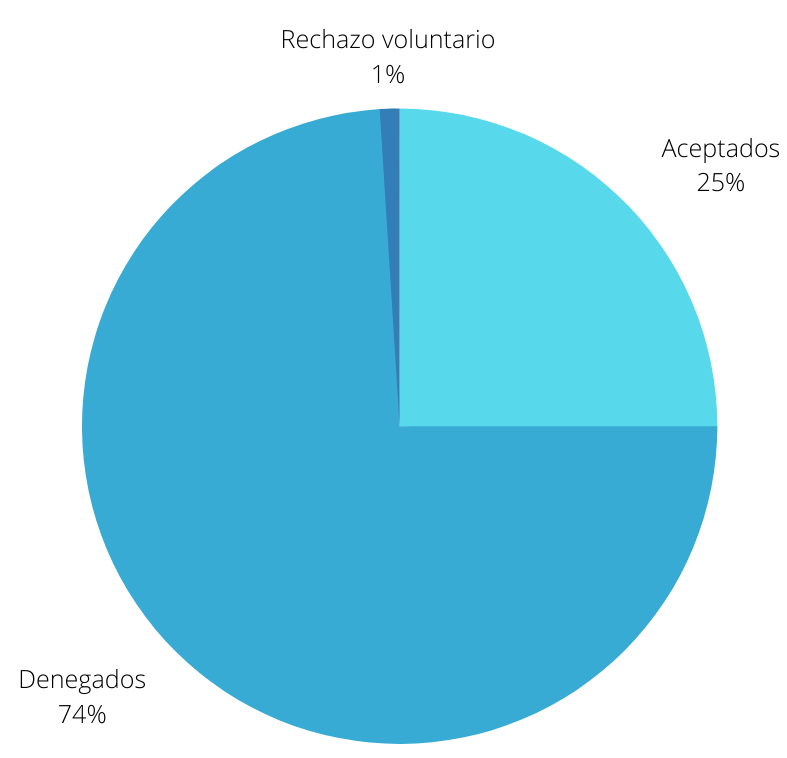
En la votación realizada sobre la concesión, de la autorización se obtuvieron un total de 6 votos a favor, 15 votos en contra, y 12 votos en blanco, dividiéndose éstos en preguntas como:
- ¿El Estudio 302, visto de forma independiente y sin tener en cuenta el Estudio 301, proporcionar pruebas sólidas que respalden la eficacia de aducanumab para la tratamiento de la enfermedad de Alzheimer? Esta pregunta obtuvo un total de un voto a favor, 8 en contra y 2 en blanco.
- ¿El estudio 103 proporciona evidencia de apoyo de la efectividad de aducanumab para el tratamiento de la enfermedad de Alzheimer? Esta pregunta obtuvo 0 votos a favor, 7 en contra y 4 en blanco.
- ¿Ha presentado el solicitante pruebas sólidas de un efecto farmacodinámico sobre la fisiopatología de la enfermedad de Alzheimer? En esta pregunta se obtuvieron 5 votos a favor, 0 en contra y 6 en blanco.
- ¿Considera el estudio 302 como evidencia principal de la eficacia de aducanumab para el tratamiento de la enfermedad de Alzheimer? En esta pregunta se obtuvieron 6 votos a favor, 15 en contra y 12 en blanco.
 Fuente: una comparación de aducanumab con otros mAbs muestra que mientras que los últimos mAb se dirigieron en gran medida contra la región del extremo amino de la péptido Aβ y solo reconocieron monómeros, aducanumab se dirigió contra un epítopo conformacional que se encuentra únicamente en los oligómeros del péptido Aβ tóxico.
Fuente: una comparación de aducanumab con otros mAbs muestra que mientras que los últimos mAb se dirigieron en gran medida contra la región del extremo amino de la péptido Aβ y solo reconocieron monómeros, aducanumab se dirigió contra un epítopo conformacional que se encuentra únicamente en los oligómeros del péptido Aβ tóxico.
La FDA ha adjuntado en la carta en la que notificaban a Biogen la concesión de la autorización, un punto adicional que indica que deben realizar un ensayo clínico aleatorizado y controlado con el Gold Standard que ellos consideren siguiendo las siguientes fechas:
- Enviar el borrador del protocolo – 10/2021
- Enviar el protocolo final – 08/2022
- Haber finalizado el ensayo clínico – 08/2029
- Enviar el informe final de ensayo – 02/2030
En la carta, la FDA les avisa de que:
«SI LOS ENSAYOS CLÍNICOS POSTERIORES A LA COMERCIALIZACIÓN NO LOGRAN VERIFICAR EL BENEFICIO CLÍNICO O NO SE LLEVAN A CABO CON LA DEBIDA DILIGENCIA, SE PODRÁ RETIRAR ESTA APROBACIÓN»
Desde el otro lado del charco, la Agencia Europea del Medicamento (EMA) ha desaconsejado a los países miembros la autorización del aducanumab. Habrá que esperar a los resultados del ensayo clínico impuesto por la FDA, y a los informes de farmacovigilancia que se vayan emitiendo, para saber si la EMA se retractará, o no, de su decisión sobre la autorización de aducanumab en los países de los Estados miembros de la Unión Europea.
Para cualquier duda relacionada con este post puedes escribirnos aquí.